Автор: Денис Аветисян
Исследователи разработали дифференцируемую платформу, сочетающую методы машинного обучения и теорию функционала плотности для повышения точности и эффективности расчетов электронных структур периодических систем.
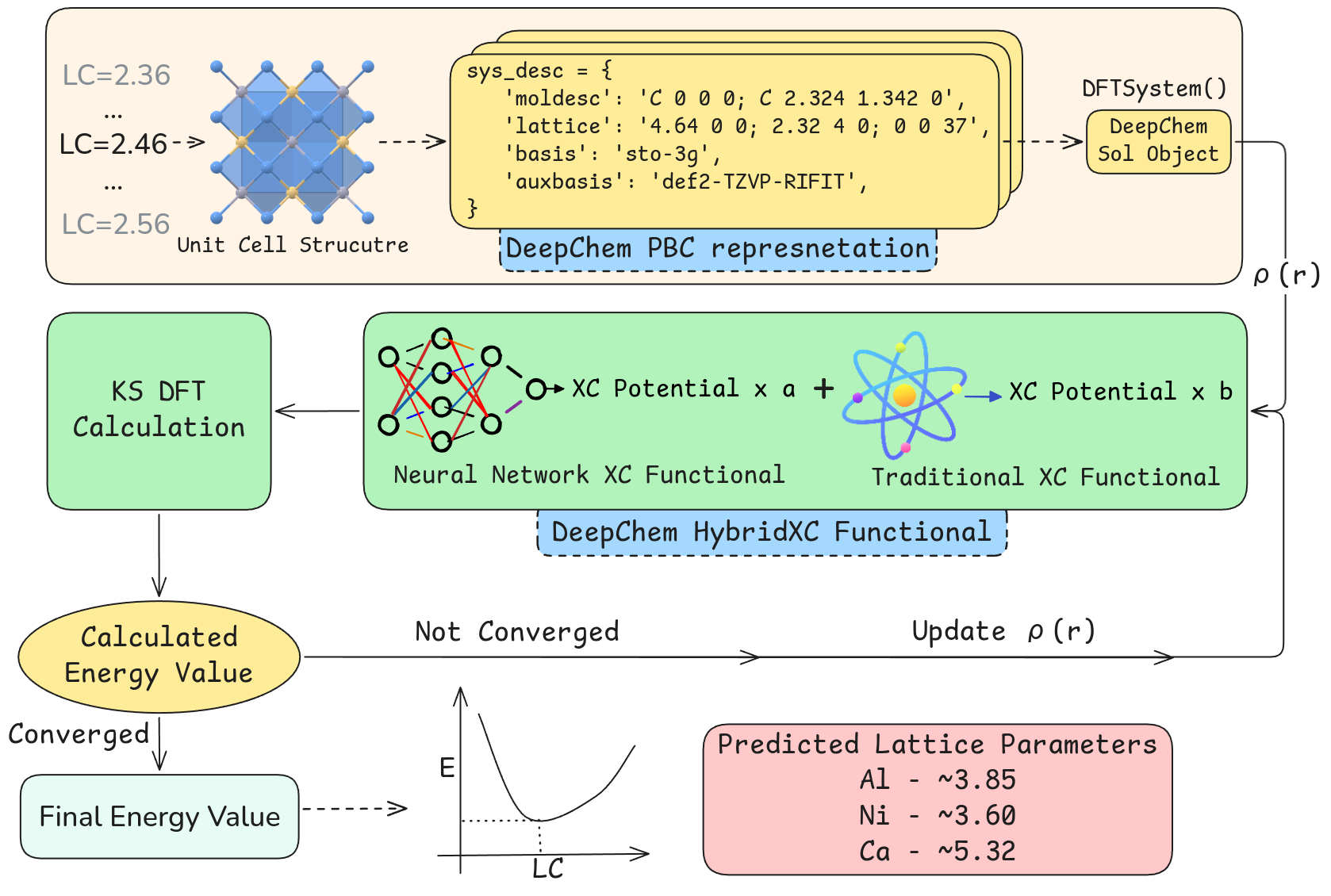
Представлен фреймворк, позволяющий обучать прокси-функционалы обмена и корреляции для кристаллических материалов с использованием дифференцируемых расчетов теории функционала плотности и нейронных сетей.
Несмотря на широкое распространение теории функционала плотности (DFT) в материаловедении и химии, вычислительные затраты остаются существенным ограничением при моделировании крупных систем. В данной работе, посвященной разработке ‘A fully differentiable framework for training proxy Exchange Correlation Functionals for periodic systems’, представлен дифференцируемый фреймворк, интегрирующий модели машинного обучения в DFT для кристаллических материалов и периодических систем. Предложенный подход позволяет обучать аппроксимации функционалов обмена и корреляции, используя нейронные сети и обеспечивая возможность вычисления градиентов через весь самосогласованный процесс DFT. Позволит ли это создать более точные и эффективные методы электронно-структурных расчетов, расширяя горизонты моделирования сложных материалов?
Вычислительные Пределы и Поиск Новых Материалов
Точное моделирование материалов базируется на теории функционала плотности (DFT), однако её применение сопряжено со значительными вычислительными затратами. Этот метод, требующий решения сложных уравнений для описания поведения электронов в материале, ограничивает масштаб симуляций и препятствует исследованию систем, состоящих из большого числа атомов. Чем сложнее материал и чем точнее требуется результат, тем больше вычислительных ресурсов необходимо. Например, моделирование дефектов, поверхностей или динамических процессов требует значительно больших затрат, чем анализ идеальной кристаллической решетки. В результате, возможность предсказать свойства новых материалов или оптимизировать существующие часто ограничена вычислительной мощностью доступного оборудования, что создает серьезное препятствие для прогресса в материаловедении и смежных областях.
Традиционные методы теории функционала плотности (DFT) сталкиваются со значительными трудностями при точном описании электронных корреляций — взаимодействия между электронами, выходящего за рамки независимой модели электронов. Это связано с тем, что точное вычисление этих взаимодействий требует экспоненциального увеличения вычислительных ресурсов с ростом числа электронов в системе. В результате, упрощенные приближения, используемые в стандартных DFT-вычислениях, приводят к неточностям в предсказании свойств материалов, особенно в случаях, когда электронные корреляции играют доминирующую роль. Компенсация этих неточностей требует существенного увеличения вычислительных затрат, ограничивая размер исследуемых систем и количество проведенных расчетов. Поэтому, разработка более эффективных методов учета электронных корреляций является ключевой задачей для продвижения моделирования материалов и открытия новых веществ с заданными свойствами.
Поиск новых материалов с заданными свойствами сталкивается с серьезными ограничениями, обусловленными непомерной вычислительной стоимостью исчерпывающего скрининга на основе теории функционала плотности (DFT). Традиционный подход, требующий проведения DFT-расчетов для огромного числа потенциальных соединений, становится практически неосуществимым даже при использовании самых мощных суперкомпьютеров. Это связано с экспоненциальным ростом вычислительных затрат с увеличением числа атомов в моделируемой системе и необходимостью точного учета сложных взаимодействий между электронами. В результате, исследователи вынуждены ограничиваться изучением лишь небольшой части потенциального материального пространства, что значительно замедляет процесс открытия и разработки инновационных материалов с улучшенными характеристиками. Поэтому, преодоление этой вычислительной преграды является ключевой задачей для развития материаловедения и создания материалов будущего.
В настоящее время, машинное обучение выступает в качестве перспективного инструмента для преодоления вычислительных ограничений в материаловедении. Вместо проведения дорогостоящих расчетов на основе теории функционала плотности (DFT), создаются так называемые суррогатные модели. Эти модели, обученные на относительно небольшом наборе данных, полученных с помощью DFT, способны предсказывать свойства материалов с сопоставимой точностью, но в сотни или даже тысячи раз быстрее. Такой подход позволяет значительно ускорить процесс поиска новых материалов с заданными характеристиками, открывая возможности для масштабного скрининга и виртуальных экспериментов, которые ранее были практически невозможны из-за ограничений вычислительных ресурсов. Разработка и применение этих суррогатных моделей — ключевой шаг к автоматизации и ускорению материаловедческих исследований.
Глубокое Обучение для Ускорения Электронной Структуры
Глубокое обучение, в частности, с использованием фреймворков DeepChem и PyTorch, предоставляет мощную платформу для создания машинных моделей-заменителей (суррогатных моделей). Эти модели позволяют аппроксимировать сложные вычисления, обычно требующие значительных вычислительных ресурсов, за счет обучения на больших наборах данных. Фреймворк DeepChem предоставляет специализированные инструменты для работы с данными, характерными для химии и материаловедения, а PyTorch обеспечивает гибкость и возможности для кастомизации архитектур нейронных сетей. Использование суррогатных моделей позволяет значительно ускорить расчеты электронных свойств материалов, делая возможным исследования, которые ранее были практически невозможны из-за вычислительных ограничений. Такие модели могут быть обучены для предсказания энергии, сил и других свойств, необходимых для моделирования материалов.
Нейронные функционалы обмена-корреляции, обученные на данных, полученных с помощью теории функционала плотности (DFT), позволяют выявлять сложные зависимости между атомной структурой и электронными свойствами материалов. Эти модели используют глубокие нейронные сети для аппроксимации функционала обмена-корреляции, который определяет энергию основного состояния системы. Обучение проводится на обширных наборах данных, генерируемых высокоточными DFT расчетами, позволяя модели «выучить» поведение электронов в различных материалах. В отличие от традиционных аппроксимаций, нейронные сети способны захватывать нелинейные и многомерные корреляции, что потенциально приводит к более точным предсказаниям энергетических свойств, включая энергии связей, энергии активации и плотность состояний. Точность модели напрямую зависит от качества и объема обучающих данных, а также от архитектуры и параметров используемой нейронной сети.
Успех моделей машинного обучения для расчетов электронной структуры твердых тел напрямую зависит от адекватного представления их структуры. Класс Sol в рамках библиотеки DeepChem предоставляет специализированную инфраструктуру для работы с твердыми телами, позволяя эффективно кодировать информацию о кристаллической решетке, типах атомов и их координации. Это включает в себя поддержку различных форматов файлов, содержащих информацию о кристаллической структуре, а также инструменты для обработки и преобразования этих данных в формат, пригодный для обучения нейронных сетей. В частности, Sol предоставляет средства для генерации дескрипторов, описывающих локальное окружение каждого атома в кристаллической структуре, что позволяет моделировать сложные зависимости между структурой и электронными свойствами материала.
В основе обучения суррогатных моделей, используемых в глубоком обучении для ускорения расчетов электронной структуры, лежит метод функционала плотности (DFT) Kohn-Sham. Этот метод служит генератором высокоточных данных, необходимых для обучения модели. DFT Kohn-Sham позволяет рассчитывать электронную структуру материалов с высокой степенью достоверности, предоставляя эталонные значения, которые используются для тренировки нейронных сетей. Объем и качество данных, полученных с помощью DFT, напрямую влияют на точность и обобщающую способность суррогатных моделей, позволяя им эффективно предсказывать электронные свойства новых материалов и структур. На практике, расчеты DFT используются для создания больших наборов данных, охватывающих различные типы материалов и структурные конфигурации, которые затем служат основой для обучения моделей машинного обучения.
Гибридные Функционалы: Сочетание Точности и Эффективности
Класс HybridXC объединяет в себе преимущества традиционных функционалов обобщенного градиентного приближения (GGA) с выразительной мощностью многослойных перцептронов. Функционалы GGA обеспечивают вычислительную эффективность, однако могут быть недостаточно точными в описании сложных электронных систем. Использование многослойных перцептронов позволяет обучать модель для внесения корректировок к GGA функционалу, тем самым повышая точность предсказаний без значительного увеличения вычислительных затрат. По сути, HybridXC использует преимущества обоих подходов, сочетая скорость GGA с улучшенной точностью, обеспечиваемой машинным обучением.
Гибридный подход позволяет модели обучаться и вносить корректировки к функционалам обобщенного градиентного приближения (GGA), повышая точность расчетов без значительного увеличения вычислительных затрат. Вместо полного пересчета электронных свойств, модель изучает разницу между результатами GGA и более точными, но ресурсоемкими расчетами, полученными, например, с использованием методов DFT высокого уровня. Это позволяет предсказывать свойства материалов с большей точностью, сохраняя при этом сравнимую скорость вычислений с традиционными GGA функционалами. Фактически, модель «уточняет» GGA функционал, добавляя к нему обучаемые параметры, которые компенсируют систематические ошибки.
Для эффективного обучения моделей HybridXC требуется тщательно подготовленный набор данных. Генерация этого набора данных осуществляется методами теории функционала плотности (DFT) Kohn-Sham, используя минимальный базисный набор STO-3G и обобщенный функционал обмена-корреляции PBE. Использование STO-3G позволяет снизить вычислительные затраты на этапе генерации данных, что важно при создании больших обучающих наборов. PBE функционал выбран как надежный и широко используемый, обеспечивающий достаточную точность для генерации данных, используемых для обучения модели, корректирующей GGA функционалы.
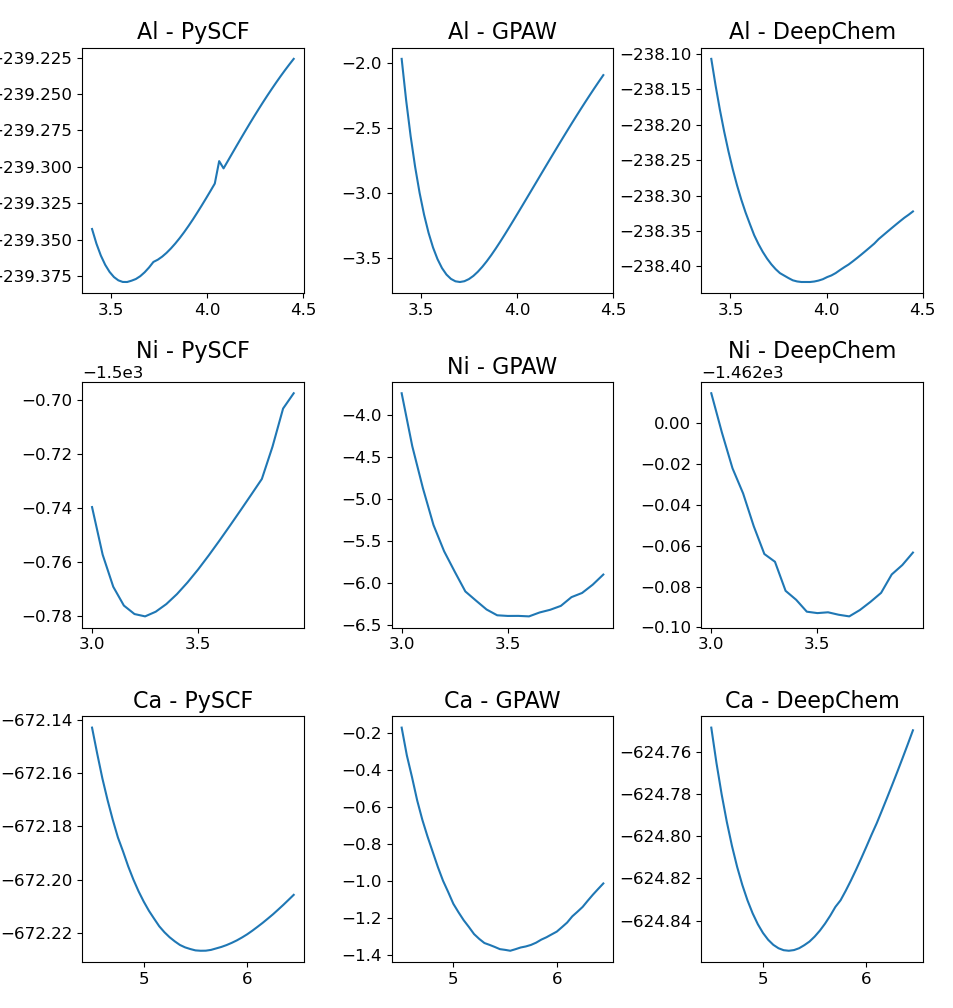
Валидация функционалов HybridXC проводится посредством оптимизации констант решетки для систем с гранецентрированной кубической структурой. Результаты демонстрируют относительную погрешность в предсказании констант решетки, составляющую приблизительно 5-10% по сравнению с эталонными расчетами DFT, выполненными с использованием общепринятых пакетов программного обеспечения, таких как GPAW и PySCF. Данная оценка погрешности позволяет оценить точность и надежность предсказаний, полученных с использованием разработанных функционалов, в контексте структурных свойств материалов.
Будущее Развитие: Масштабируемость и За Ее Пределами
Проект «Materials Project» представляет собой ценнейший источник экспериментальных данных, служащий основой для дальнейшего обучения и верификации разработанной модели. Обширная база, включающая проверенные сведения о структуре, свойствах и синтезе различных материалов, позволяет значительно повысить точность и надежность предсказаний. Использование этих данных в качестве эталона при обучении алгоритмов машинного обучения способствует созданию более реалистичных и применимых моделей, способных эффективно прогнозировать характеристики новых материалов и оптимизировать существующие. Постоянное расширение и обновление базы данных «Materials Project» обеспечивает непрерывное улучшение модели и расширение ее возможностей в области материаловедения и химии.
Переход от гауссовых базисных наборов к плоским волнам обещает значительно расширить возможности модели при работе с крупномасштабными системами и сложными материалами. Гауссовы функции, хоть и эффективны для описания локализованных электронных состояний, становятся вычислительно затратными при моделировании периодических структур и больших ячеек. Плоские волны, напротив, естественным образом описывают периодичность кристаллов и обладают более предсказуемой вычислительной сложностью, масштабирующейся линейно с размером системы. Это позволяет исследовать материалы с более реалистичными размерами и дефектами, а также моделировать процессы, происходящие на больших временных масштабах. Использование плоских волн открывает путь к проведению высокоточных расчетов для новых материалов, обладающих уникальными свойствами и потенциально востребованных в различных областях науки и техники.
Предложенная методика открывает принципиально новые возможности для ускорения открытия материалов. Благодаря возможности быстро и эффективно оценивать свойства огромного числа химических соединений, становится возможным высокопроизводительный скрининг — просеивание обширных химических пространств с целью выявления наиболее перспективных кандидатов для дальнейших исследований. Такой подход позволяет значительно сократить время и ресурсы, необходимые для разработки новых материалов с заданными характеристиками, например, для повышения эффективности солнечных батарей, создания более прочных сплавов или разработки новых катализаторов. Вместо традиционных, трудоемких методов, требующих проведения большого количества физических экспериментов или сложных компьютерных симуляций для каждого материала, предложенный фреймворк позволяет оценить потенциал тысяч, а возможно и миллионов соединений, значительно расширяя горизонты материаловедения.
Снижение вычислительных затрат при моделировании материалов открывает принципиально новые возможности для создания веществ с заданными характеристиками, востребованными в самых различных областях. Это позволяет перейти от традиционного, часто эмпирического подхода к материаловедению, к более точному и предсказуемому проектированию. Например, можно разрабатывать новые высокоэффективные катализаторы для химической промышленности, сверхпроводящие материалы для передачи энергии без потерь, или легкие и прочные композиты для авиакосмической отрасли. Уменьшение времени и ресурсов, необходимых для моделирования, стимулирует инновации и ускоряет процесс открытия материалов с уникальными свойствами, что потенциально может привести к технологическому прорыву в энергетике, медицине и других ключевых сферах.
Представленная работа демонстрирует отказ от жесткого контроля над функционалами обмена и корреляции в рамках теории функционала плотности. Вместо этого, предлагается гибкий, обучаемый подход, где поправки к существующим функционалам определяются на основе данных. Это перекликается с идеей о том, что система — это живой организм, где локальные взаимодействия и адаптация важнее централизованного управления. Как заметил Пол Фейерабенд: «В науке нет никаких универсальных правил, только полезные хитрости». Данное исследование, позволяющее функционалам эволюционировать и адаптироваться к конкретным кристаллическим структурам, подтверждает эту мысль — эффективность возникает не из строгих предписаний, а из гибкого реагирования на локальные условия, что особенно важно при моделировании периодических систем.
Что дальше?
Представленная работа, хотя и демонстрирует элегантное слияние дифференцируемого функционального анализа и машинного обучения, лишь приоткрывает завесу над истинным потенциалом самоорганизующихся систем в рамках теории функционала плотности. Попытки «обучить» функционалы обходятся дорого, и акцент на корректировках существующих, пусть и удобен, не устраняет фундаментальную неопределенность в описании электронного обмена и корреляции. Каждая точка связи между атомами в кристаллической решетке несет влияние, и, возможно, более продуктивным путем окажется не поиск «лучшего» функционала, а позволение системе самой находить оптимальное описание, используя локальные правила, а не централизованный контроль.
Очевидным ограничением остается вычислительная стоимость, особенно при работе с большими системами и сложными функционалами. Однако, истинная проблема не в скорости вычислений, а в вере в возможность полного контроля над сложными процессами. Поиск универсального решения — иллюзия. Вместо этого, следует сосредоточиться на разработке адаптивных схем, которые позволяют системе динамически подстраиваться к конкретным условиям, используя принципы самоорганизации.
Будущие исследования, вероятно, направятся в сторону разработки более эффективных архитектур нейронных сетей, способных улавливать сложные корреляции между электронами. Но куда более перспективным представляется отказ от представления об «оптимальном» функционале вообще. Реальное управление заключается не в контроле, а во влиянии — создании условий, в которых система сама найдет равновесие, соответствующее наблюдаемым свойствам материала.
Оригинал статьи: https://arxiv.org/pdf/2602.15923.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- ПРОГНОЗ ДОЛЛАРА К ШЕКЕЛЮ
- БИТКОИН ПРОГНОЗ. BTC криптовалюта
- SIREN ПРОГНОЗ. SIREN криптовалюта
- MYX ПРОГНОЗ. MYX криптовалюта
- ЭФИРИУМ ПРОГНОЗ. ETH криптовалюта
- SAROS ПРОГНОЗ. SAROS криптовалюта
- SOL ПРОГНОЗ. SOL криптовалюта
- ZEC ПРОГНОЗ. ZEC криптовалюта
- ПРОГНОЗ ДОЛЛАРА
- ДОГЕКОИН ПРОГНОЗ. DOGE криптовалюта
2026-02-19 21:01