Автор: Денис Аветисян
Новый подход с использованием графовых нейронных сетей позволяет быстро и точно предсказывать инфракрасные спектры полициклических ароматических углеводородов, ключевых компонентов межзвездной пыли.
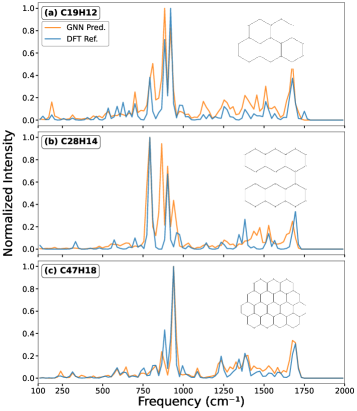
Исследование демонстрирует эффективность архитектуры Attentive Fingerprint для прогнозирования инфракрасных спектров ПАУ, значительно превосходящую традиционные квантово-химические расчеты по скорости, хотя экстраполяция к очень крупным молекулам остается сложной задачей.
Анализ ароматических инфракрасных полос (AIB), наблюдаемых в межзвездном пространстве, затруднен из-за структурного разнообразия полициклических ароматических углеводородов (ПАУ). В работе, посвященной ‘Graph Neural Network Prediction of Infrared Spectra of Interstellar Polycyclic Aromatic Hydrocarbons’, разработан эффективный фреймворк на основе графовых нейронных сетей (ГНС) для предсказания инфракрасных спектров ПАУ, превосходящий по скорости традиционные квантово-химические расчеты. Наилучшая производительность продемонстрирована архитектурой Attentive Fingerprint (AFP) с использованием расхождения Дженсена-Шеннона в качестве функции потерь. Сможет ли данный подход существенно ускорить анализ AIB и расширить наше понимание межзвездной среды, особенно при изучении более крупных молекул ПАУ?
Неразгаданные инфракрасные сигналы: отголоски звёздной химии
В астрономических спектрах повсеместно обнаруживаются неидентифицированные инфракрасные эмиссионные линии, известные как UIE (Unidentified Infrared Emission). Эти загадочные сигналы свидетельствуют о существовании сложных молекулярных структур в межзвездном пространстве. Их широкое распространение в различных галактиках и областях звездообразования указывает на то, что эти молекулы играют важную роль в космической химии и физике. Хотя точная природа этих эмиссионных линий остается предметом исследований, их характеристики позволяют предположить наличие полициклических ароматических углеводородов (ПАУ) и других сложных органических соединений, которые могут формироваться в результате процессов, происходящих в звездных недрах и в межзвездной среде. Изучение UIE предоставляет уникальную возможность понять состав и эволюцию межзвездной среды, а также раскрыть механизмы формирования сложных молекул во Вселенной.
Полициклические ароматические углеводороды (ПАУ) считаются наиболее вероятными носителями неидентифицированного инфракрасного излучения, наблюдаемого в космосе. Однако, чтобы подтвердить эту роль, необходимы точные модели их спектров. Сложность заключается в том, что спектральные характеристики ПАУ чрезвычайно чувствительны к их размеру, структуре и химическим модификациям. Поэтому, для адекватного сопоставления с астрономическими наблюдениями, требуется учитывать огромное разнообразие молекулярных конфигураций и точно воспроизводить их инфракрасные сигнатуры. Успешное моделирование позволит не только подтвердить присутствие ПАУ в межзвездной среде, но и получить ценную информацию об их происхождении, эволюции и роли в космической химии.
Традиционные методы расчета спектров полициклических ароматических углеводородов (ПАУ) сталкиваются с серьезными вычислительными ограничениями. Сложность этих расчетов возрастает непропорционально размеру молекулы, а именно — с зависимостью порядка N_C^{4.18}, где N_C обозначает число атомов углерода в молекуле ПАУ. Это означает, что даже умеренное увеличение размера молекулы приводит к экспоненциальному росту требуемых вычислительных ресурсов, делая анализ больших ПАУ, потенциально важных для понимания межзвездной среды, практически невозможным. В результате, существующие модели часто ограничены анализом относительно небольших молекул, что может приводить к неполному или искаженному представлению о реальных спектральных особенностях, наблюдаемых в астрономических спектрах.
От отпечатков пальцев к машинному предсказанию спектров
Ранние подходы машинного обучения (ML) в изучении полициклических ароматических углеводородов (ПАУ) широко использовали молекулярные отпечатки (Molecular Fingerprints) в качестве упрощенного представления структуры молекул. Эти отпечатки, по сути, являются бинарными векторами, кодирующими наличие или отсутствие определенных структурных фрагментов в молекуле ПАУ. Использование молекулярных отпечатков позволило преобразовать сложные химические структуры в числовой формат, пригодный для обработки алгоритмами машинного обучения, такими как случайные леса и нейронные сети. Хотя молекулярные отпечатки обеспечивают компактное представление, они неизбежно теряют информацию о трехмерной структуре и деталях электронного строения молекулы ПАУ, что ограничивает точность прогнозирования спектральных характеристик.
Модели машинного обучения, такие как случайный лес (Random Forest) и нейронные сети прямого распространения (Feedforward Neural Networks), показали возможность установления связи между структурой полициклических ароматических углеводородов (ПАУ) и их спектрами. В ходе исследований было продемонстрировано, что эти модели способны обучаться на данных о структуре молекул ПАУ и предсказывать соответствующие им спектральные характеристики. Достигнутая корреляция между входными данными, представляющими структуру молекулы, и выходными данными, представляющими спектр, подтверждает принципиальную возможность использования методов машинного обучения для прогнозирования спектров ПАУ на основе их структурных особенностей.
Несмотря на продемонстрированную возможность обучения моделей, таких как случайный лес и нейронные сети, установлению взаимосвязи между структурой и спектром ПАУ, эти методы испытывали трудности при полном воспроизведении тонких спектральных особенностей, возникающих в сложных молекулах ПАУ. Ограничения были связаны с упрощенным представлением структуры, используемым в молекулярных отпечатках, которое не всегда позволяло адекватно отразить все факторы, влияющие на спектральные характеристики, особенно в случае больших или функционализированных ПАУ. Это приводило к неточностям в предсказании спектров и снижало эффективность моделей при анализе реальных астрономических данных.
База данных ПАУ, разработанная в Лаборатории Эймса NASA с использованием теории функционала плотности (DFT), предоставила критически важные обучающие данные для моделей машинного обучения. DFT-расчеты позволили получить точные предсказания электронных спектров и других свойств широкого спектра полициклических ароматических углеводородов (ПАУ). Эта база данных содержит информацию о геометрии молекул, энергетических уровнях и вероятностях переходов, что позволило создать надежные наборы данных для обучения и валидации алгоритмов, таких как случайные леса и нейронные сети. Включение экспериментально подтвержденных данных, полученных с использованием спектроскопии, также повысило точность и применимость моделей для прогнозирования спектров ПАУ в астрофизических средах.
Графовые нейронные сети: новый взгляд на спектральный анализ
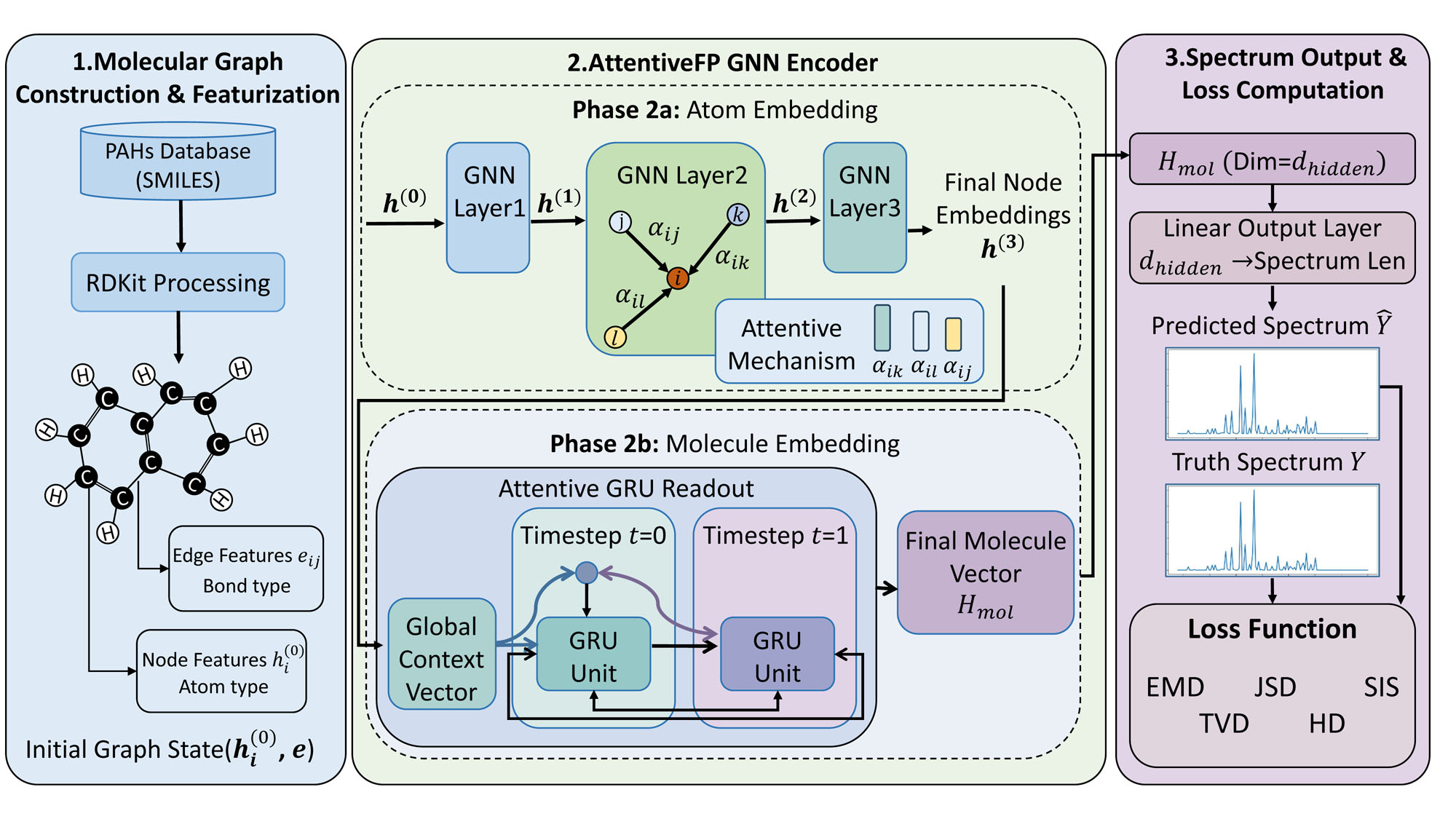
Графовые нейронные сети (GNN) представляют молекулы в виде графов, где атомы выступают в роли узлов, а химические связи — в роли ребер. Такой подход позволяет напрямую кодировать структурную связность молекулы, что существенно улучшает точность предсказания её спектральных характеристик. В отличие от традиционных методов, которые используют векторные представления молекул, GNN учитывают топологию молекулярной структуры, позволяя моделировать взаимодействия между атомами и предсказывать спектральные свойства, такие как частоты колебаний и поглощения. Это особенно важно для сложных молекул, где структурные особенности оказывают значительное влияние на спектр.
Семейство графовых нейронных сетей (GNN), включающее в себя такие архитектуры как Graph Convolutional Network (GCN), Message Passing Neural Network (MPNN) и Graph Attention Network (GAT), использует графовые представления молекул для извлечения сложных спектральных характеристик. GCN применяет свертки непосредственно на графовой структуре, агрегируя информацию от соседних узлов. MPNN обобщает этот подход, определяя фазы передачи сообщений и обновления узлов. GAT использует механизмы внимания для взвешивания вкладов соседних узлов, позволяя модели сосредотачиваться на наиболее релевантных участках молекулярного графа. Все эти модели эффективно кодируют структурные связи, позволяя им предсказывать спектральные свойства молекул с высокой точностью, превосходя традиционные методы, основанные на дескрипторах.
Для ввода молекулярных структур в архитектуры графовых нейронных сетей (GNN) широко используются SMILES-строки (Simplified Molecular Input Line Entry System). SMILES представляет собой линейную нотацию, кодирующую структуру молекулы в виде строки символов, описывающих атомы и связи между ними. Данный формат позволяет преобразовывать химическую структуру в машиночитаемый вид, необходимый для обработки GNN. Процесс включает парсинг SMILES-строки для создания графового представления молекулы, где атомы выступают в роли узлов, а химические связи — в роли ребер. Это позволяет GNN эффективно обрабатывать информацию о молекулярной структуре и использовать ее для задач, таких как предсказание спектральных свойств.
Архитектура Attentive Fingerprint, являясь разновидностью графовой нейронной сети (GNN), использует механизмы внимания (attention) для повышения точности предсказания спектров молекул. В отличие от стандартных GNN, где информация о каждом атоме распространяется одинаково по графу, Attentive Fingerprint динамически взвешивает вклад каждого соседнего атома при вычислении представления текущего атома. Это достигается путем обучения весов внимания, которые определяют значимость каждого соседа в контексте конкретного атома и предсказываемого спектрального свойства. Использование механизмов внимания позволяет модели фокусироваться на наиболее релевантных структурных особенностях молекулы, что приводит к более точным и надежным предсказаниям спектров по сравнению с традиционными подходами.
Оценка спектрального сходства с помощью передовых метрик
Для точного прогнозирования спектров необходимо использовать надежные метрики расстояния, которые позволяют количественно оценить сходство между предсказанным и наблюдаемым спектрами. Эти метрики служат для определения степени различия между двумя спектральными распределениями, учитывая интенсивность излучения на различных длинах волн. Выбор подходящей метрики критически важен, поскольку он напрямую влияет на точность оценки расхождений и, следовательно, на интерпретацию полученных результатов. Например, небольшие различия в форме спектра могут указывать на незначительные изменения в составе или физических условиях исследуемого объекта, в то время как существенные отклонения могут свидетельствовать о принципиально иных процессах. Эффективность алгоритмов, использующих прогнозирование спектров, напрямую зависит от корректности и чувствительности выбранных метрик расстояния.
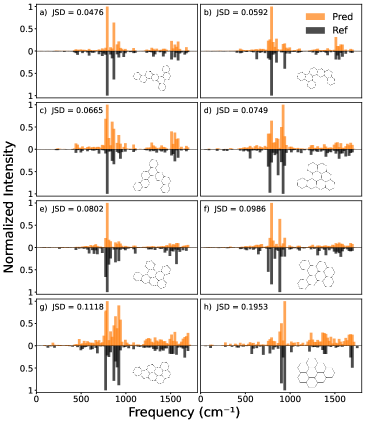
Для количественной оценки схожести предсказанных и наблюдаемых спектров используются различные метрики расстояния, включая расстояние Землекопа (Earth Mover’s Distance), расхождение Дженсена-Шеннона (Jensen-Shannon Divergence), расстояние Хеллингера (Hellinger Distance), расстояние полной вариации (Total Variation Distance) и сходство спектральной информации (Spectrum Information Similarity). Каждая из этих метрик имеет свои особенности в расчете различий между распределениями, что позволяет оценить степень соответствия между предсказанным и наблюдаемым спектрами. Расстояние Землекопа, например, учитывает «стоимость» перемещения одного распределения в другое, в то время как расхождение Дженсена-Шеннона измеряет разницу между двумя вероятностными распределениями. Выбор конкретной метрики зависит от характеристик анализируемых спектров и целей исследования.
В архитектуре Attentive Fingerprint указанные метрики расстояний (Earth Mover’s Distance, Jensen-Shannon Divergence, Hellinger Distance, Total Variation Distance и Spectrum Information Similarity) используются в качестве функций потерь при обучении нейронной сети. Это означает, что в процессе оптимизации сеть стремится минимизировать значение выбранной метрики между предсказанным и наблюдаемым спектрами. Использование этих метрик в качестве функций потерь позволяет сети не просто приближенно воспроизводить спектры, но и учитывать их тонкие особенности, что критически важно для точной идентификации и характеристики полициклических ароматических углеводородов (PAH) в астрономических наблюдениях. Выбор конкретной метрики влияет на чувствительность сети к различным типам спектральных расхождений и, следовательно, на качество получаемых результатов.
Эффективность используемых метрик спектрального сходства напрямую влияет на точность идентификации и характеристики полициклических ароматических углеводородов (ПАУ) в астрономических наблюдениях. Различные метрики, такие как расстояние Землеройки (Earth Mover’s Distance), расхождение Дженсена-Шеннона, расстояние Хеллингера, расстояние полной вариации и сходство спектральной информации, позволяют количественно оценить различия между предсказанными и наблюдаемыми спектрами ПАУ. Более точная оценка этих различий, обеспечиваемая оптимально подобранной метрикой, критически важна для корректной интерпретации астрономических данных и получения достоверной информации о составе и свойствах межзвездной среды, где ПАУ играют значительную роль.
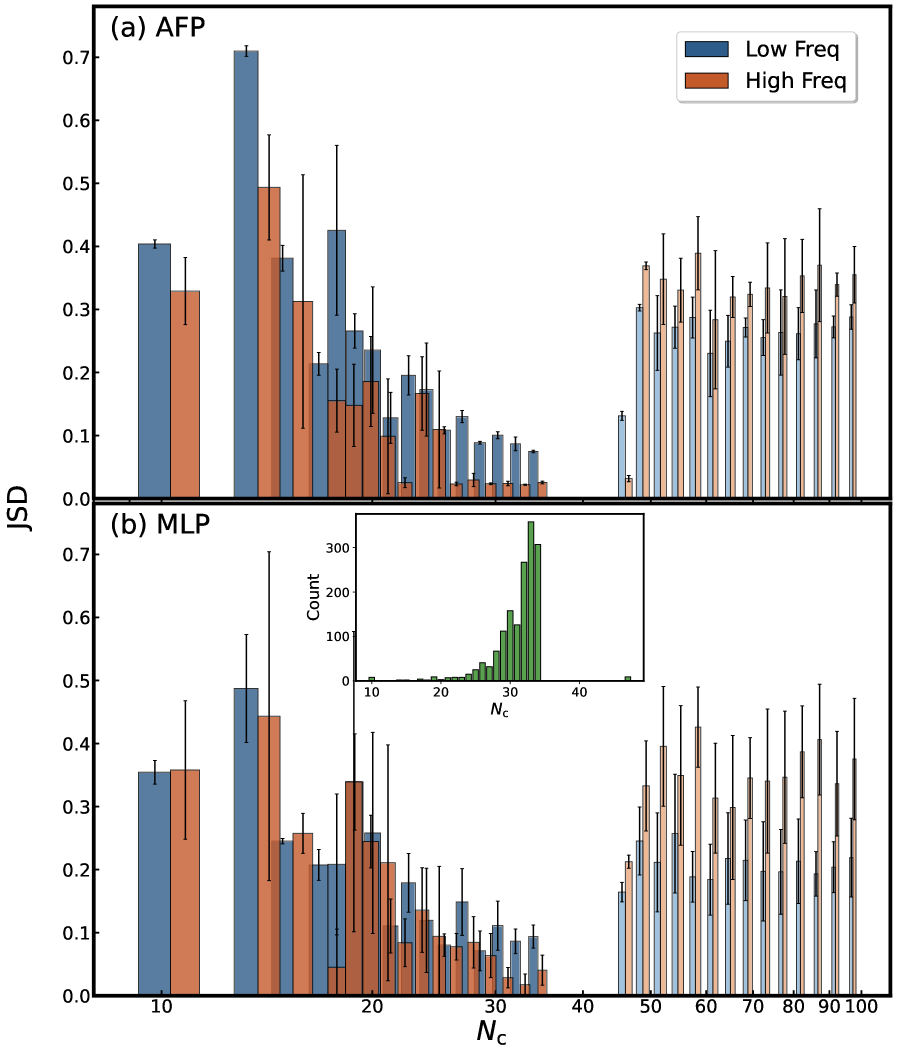
К более глубокому пониманию межзвездной среды
Интеграция графовых нейронных сетей (GNN) и передовых спектральных метрик знаменует собой существенный прогресс в моделировании спектров полициклических ароматических углеводородов (ПАУ). Традиционные методы, такие как расчеты в рамках теории функционала плотности, оказываются вычислительно затратными, особенно при работе со сложными молекулами ПАУ. Применение GNN позволяет эффективно кодировать структурную информацию молекул и устанавливать взаимосвязь между структурой и спектральными характеристиками. Сочетание этой способности с усовершенствованными спектральными метриками позволяет получать более точные и быстрые прогнозы спектров ПАУ, что открывает новые возможности для анализа астрономических данных и понимания состава межзвездной среды. Этот подход позволяет исследователям более эффективно идентифицировать и количественно оценивать ПАУ в различных астрофизических условиях, расширяя наши знания об их роли в формировании и эволюции галактик.
Улучшенные возможности моделирования имеют решающее значение для интерпретации характеристик UIE (Unidentified Infrared Emission) — загадочных инфракрасных излучений, наблюдаемых в межзвездной и околозвездной среде. Эти особенности спектра, долгое время остававшиеся неразгаданными, предположительно связаны с полициклическими ароматическими углеводородами (ПАУ), но точное определение их структуры и концентрации представляет собой сложную задачу. Более точное моделирование позволяет ученым сопоставлять наблюдаемые спектры UIE с теоретическими предсказаниями, что, в свою очередь, дает возможность оценить количество и тип ПАУ, присутствующих в этих космических средах. Это понимание крайне важно для изучения состава, эволюции и физических условий межзвездной среды, а также для раскрытия процессов формирования звезд и планет.
Точное выявление и количественная оценка полициклических ароматических углеводородов (ПАУ) открывает новые возможности для понимания состава и эволюции межзвездной среды. ПАУ, являясь одними из наиболее распространенных органических молекул во Вселенной, играют ключевую роль в процессах формирования звезд и планет, а также в химической эволюции галактик. Их концентрация и структура служат своеобразными “отпечатками пальцев”, позволяющими реконструировать физические условия в межзвездных облаках, такие как температура, плотность и интенсивность ультрафиолетового излучения. Анализ спектральных характеристик ПАУ предоставляет ценную информацию о процессах их образования, разрушения и модификации в различных астрофизических средах, способствуя более глубокому пониманию сложных химических процессов, протекающих в космосе.
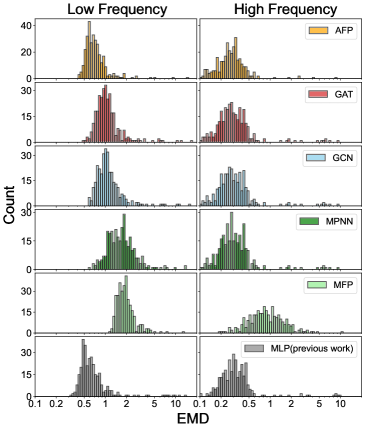
Разработанная модель Attentive Fingerprint (AFP) демонстрирует существенный прорыв в скорости и эффективности моделирования полициклических ароматических углеводородов (ПАУ). В отличие от традиционных расчетов на основе теории функционала плотности (DFT), имеющих вычислительную сложность, масштабирующуюся как N_C^{4.18}, AFP достигает ускорения в 2-5 порядков, а её сложность составляет всего N_C^{0.21}, где N_C — число атомов углерода. Проверка с использованием метрики Earth Mover’s Distance подтверждает, что AFP последовательно превосходит другие графовые нейронные сети (GNN) в точности, особенно при анализе ПАУ среднего размера (с N_C от 21 до 34), что делает её мощным инструментом для исследования межзвездной среды.

Исследование, представленное в данной работе, демонстрирует впечатляющую способность графовых нейронных сетей предсказывать инфракрасные спектры полициклических ароматических углеводородов. Подобный подход позволяет значительно ускорить процесс анализа молекулярных данных, обходя необходимость трудоёмких квантово-химических расчётов. Однако, как и любая модель, предложенная для описания сложной реальности, её точность ограничена, особенно при экстраполяции к очень большим молекулам. В связи с этим, уместно вспомнить слова Вернера Гейзенберга: «Чем точнее мы пытаемся определить одну величину, тем менее точно мы знаем другую». Эта фраза отражает фундаментальное ограничение в научном познании, применимое и к предсказательной силе даже самых передовых алгоритмов, ведь любая вероятность может быть уничтожена сложностью системы.
Что Дальше?
Представленная работа, подобно карманной чёрной дыре, заключает в себе огромный потенциал, но и неизбежные ограничения. Возможность быстрого предсказания инфракрасных спектров полициклических ароматических углеводородов с помощью графовых нейронных сетей, безусловно, является шагом вперёд. Однако, следует помнить, что любое приближение — лишь карта, а не сама территория. Экстраполяция к исключительно крупным молекулам остаётся проблемой, а значит, и граница, за которой модель перестаёт отражать реальность, всё ещё существует.
Погружение в бездну квантово-химических расчётов часто кажется необходимым, но эта работа демонстрирует, что иногда материя ведёт себя так, будто смеётся над нашими законами, и машинное обучение способно уловить эту иронию. Будущие исследования должны быть направлены не только на увеличение точности предсказаний, но и на понимание того, где и почему модель начинает «видеть» не то, что есть на самом деле.
Ведь в конечном счёте, любая теория, какой бы элегантной она ни была, может исчезнуть за горизонтом событий, уступая место новой. И задача науки — не построить вечный замок, а научиться быстро и эффективно строить новые карты, зная, что ни одна из них не будет идеальной.
Оригинал статьи: https://arxiv.org/pdf/2602.12560.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- ПРОГНОЗ ДОЛЛАРА К ШЕКЕЛЮ
- БИТКОИН ПРОГНОЗ. BTC криптовалюта
- SIREN ПРОГНОЗ. SIREN криптовалюта
- MYX ПРОГНОЗ. MYX криптовалюта
- ЭФИРИУМ ПРОГНОЗ. ETH криптовалюта
- SOL ПРОГНОЗ. SOL криптовалюта
- SAROS ПРОГНОЗ. SAROS криптовалюта
- ZEC ПРОГНОЗ. ZEC криптовалюта
- ПРОГНОЗ ДОЛЛАРА
- ДОГЕКОИН ПРОГНОЗ. DOGE криптовалюта
2026-02-16 15:41